(for naming conventions see section 3.1)
Each program is started with (at least) one command line argument, e.g.
programX programX.defin which the arguments specifies a filename, in which FORTRAN I/O units are connected to unix filenames. (See examples at specific programs). These ``def``-files are generated automatically when the standard WIEN2k scripts x, init_lapw or run_lapw are used, but may be tailored by hand for special applications. Using the option
x program -da def-file can be created without running the program. In addition each program reads/writes the following files:
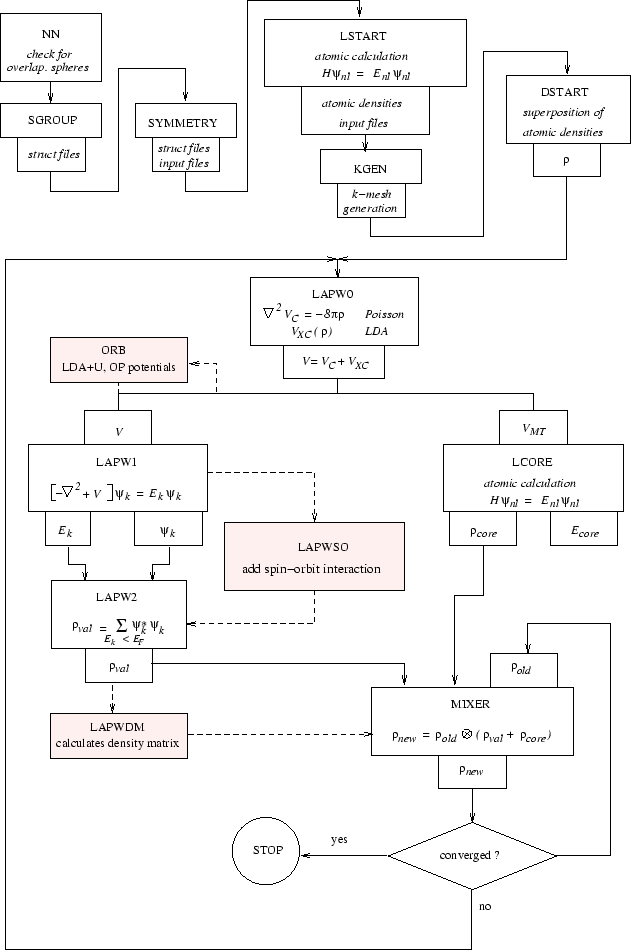
The programs of the SCF cycle (see figure 4.1) write the following files:
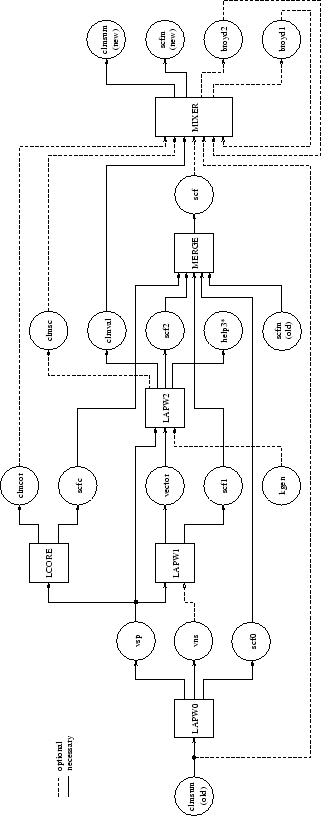 |
The following tables describe input and output files for the initialization programs nn, sgroup, symmetry, lstart, kgen, dstart (table 4.1), the utility programs tetra, irrep, spaghetti, aim, lapw7, elnes, lapw3, lapw5, xspec, optic, joint, kram, optimize and mini (table 4.2) as well as for a SCF cycle of a non-spin-polarized case (table 4.2). Optional input and output files are used only if present in the respective case subdirectory or requested/generated by an input switch. The connection between FORTRAN units and filenames are defined in the respective programX.def files. The data flow is illustrated in Fig. 4.1.
Input and output files of utility programs
| program | needs | generates | ||
| necessary | optional | necessary | optional | |
| SPAGHETTI | spaghetti.def | case.qtl | case.spaghetti_ps | case.spaghetti_ene |
| case.insp | case.outputso | case.outputsp | ||
| case.struct | case.irrep | case.band.agr | ||
| case.output1 | ||||
| TETRA | tetra.def | case.outputt | ||
| case.int | case.dos1(2,3) | |||
| case.qtl | case.dos1ev(1,2,3) | |||
| case.kgen | ||||
| LAPW3 | lapw3.def | case.output3 | ||
| case.struct | case.rho | |||
| case.in2 | ||||
| case.clmsum | case.clmsum | |||
| LAPW5 | lapw5.def | case.sigma | case.output5 | case.rho.oned |
| case.struct | case.rho | |||
| case.in5 | ||||
| case.clmval | ||||
| XSPEC | xspec.def | case.outputx | case.coredens | |
| case.inc | case.dos1ev | |||
| case.int | case.xspec | |||
| case.vsp | case.txspec | |||
| case.struct | case.m1 | |||
| case.qtl | case.m2 | |||
| OPTIC | optic.def | case.outputop | ||
| case.struct | case.symmat | |||
| case.mat_diag | ||||
| case.inop | ||||
| case.vsp | ||||
| case.vector | ||||
| JOINT | joint.def | case.outputjoint | case.sigma_intra | |
| case.injoint | case.joint | case.intra | ||
| case.struct | ||||
| case.kgen | ||||
| case.weight | ||||
| case.symmat | ||||
| case.mat_diag | ||||
| KRAM | kram.def | case.epsilon | case.eloss | |
| case.inkram | case.sigmak | case.sumrules | ||
| case.joint | ||||
| OPTIMIZE | case.struct | case_initial.struct | optimize.job | case_vol_xxxxx.struct |
| case_c/a_xxxxx.struct | ||||
| MINI | mini.def | case.scf_mini | case.outputM | case.clmsum_inter |
| case.inM | case.tmpM | case.tmpM1 | ||
| case.finM | case.constraint | case.struct1 | ||
| case.scf | case.clmhist | case.scf_mini1 | ||
| case.struct | .min_hess | .minrestart | ||
| IRREP | case.struct | case.outputirrep | ||
| case.vector | case.irrep | |||
| AIM | case.struct | case.outputaim | case.crit | |
| case.clmsum | case.surf | |||
| case.inaim | ||||
| LAPW7 | case.struct | case.output7 | case.abc | |
| case.vector | case.grid | |||
| case.in7 | case.psink | |||
| case.vsp | ||||
| QTL | case.struct | case.outputq | ||
| case.vector | case.qtl | |||
| case.inq | ||||
| case.vsp | ||||
| program | needs | generates | ||
| necessary | optional | necessary | optional | |
| LAPW0 | lapw0.def | case.clmup/dn | case.output0 | case.r2v |
| case.struct | case.vrespsum/up/dn | case.scf0 | case.vcoul | |
| case.in0 | case.inm | case.vsp(up/dn) | case.vtotal | |
| case.clmsum | case.vns(up/dn) | |||
| ORB | orb.def | case.energy | case.outputorb | case.br1orb |
| case.struct | case.vorb_old | case.scforb | case.br2orb | |
| case.inorb | case.vorb | |||
| case.dmat | orb.error | |||
| case.vsp | ||||
| LAPW1 | lapw1.def | case.vns | case.output1 | case.nsh(s) |
| case.struct | case.vorb | case.scf1 | case.nmat_only | |
| case.in1 | case.vector.old | case.vector | ||
| case.vsp | case.energy | |||
| case.klist | ||||
| LAPWSO | lapwso.def | case.vorb | case.vectorso | |
| case.struct | case.outputso | |||
| case.inso | case.scfso | |||
| case.in1 | case.energyso | |||
| case.vector | case.normso | |||
| case.vsp | ||||
| case.vns | ||||
| case.energy | ||||
| LAPW2 | lapw2.def | case.kgen | case.output2 | case.qtl |
| case.struct | case.nsh | case.scf2 | case.weight | |
| case.in2 | case.weight | case.clmval | case.weigh | |
| case.vector | case.weigh | case.help03* | ||
| case.vsp | case.recprlist | case.vrespval | ||
| case.energy | case.almblm | |||
| case.radwf | ||||
| LAPWDM | lapwdm.def | case.inso | case.outputdm | |
| case.struct | case.scfdm | |||
| case.indm | case.dmat | |||
| case.vector | lapwdm.error | |||
| case.vsp | ||||
| case.weigh | ||||
| case.energy | ||||
| SUMPARA | case.struct | case.scf2p | case.outputsum | |
| case.clmval | case.clmval | |||
| case.scf2 | ||||
| LCORE | lcore.def | case.vns | case.outputc | case.corewf |
| case.struct | case.scfc | |||
| case.inc | case.clmcor | |||
| case.vsp | lcore.error | |||
| After LCORE the case.scfX files are appended to case.scf and the | ||||
| case.clmsum file is renamed to case.clmsum_old (see run_lapw) | ||||
| MIXER | mixer.def | case.clmsum_old | case.outputm | case.broyd* |
| case.struct | case.clmsc | case.scfm | ||
| case.inm | case.clmcor | case.clmsum | ||
| case.clmval | case.scf | mixer.error | ||
| case.broyd1 | ||||
| case.broyd2 | ||||
| After MIXER the file case.scfm is appended to case.scf, so that after an iteration is | ||||
| completed, the two essential files are case.clmsum and case.scf. | ||||
In the following section the content of the (non-trivial) output files is described:
The file case.struct defines the structure and is the main input file used in all programs. We provide several examples in the subdirectory
example_struct_file
If you are using the ``Struct Generator'' from the graphical user interface w2web, you don't have to bother with this file directly! However, the description of the fields of the input mask can be found here.
Note: If you are changing this file manually, please note that this is a formatted file and the proper column positions of the characters are important! Use REPLACE instead of DELETE and INSERT during edit!
We start the description of this file with an abridged
example for rutile TiO![]() (adding line numbers):
(adding line numbers):
--------------------- top of file ---------------------line #
Titaniumdioxide TiO2 (rutile): u=0.305 1
P LATTICE,NONEQUIV. ATOMS 2 2
MODE OF CALC=RELA 3
8.6817500 8.6817500 5.5916100 90. 90. 90. 4
ATOM -1: X= 0.0000000 Y= 0.0000000 Z= 0.0000000 5
MULT= 2 ISPLIT= 8 6
ATOM -1: X= 0.5000000 Y= 0.5000000 Z= 0.5000000
Titanium NPT= 781 R0=.000022391 RMT=2.00000000 Z:22.0 7
LOCAL ROT MATRIX: -.7071068 0.7071068 0.0000000 8
0.7071068 0.7071068 0.0000000 9
0.0000000 0.0000000 1.0000000 10
ATOM -2: X= 0.3050000 Y= 0.3050000 Z= 0.0000000
MULT= 4 ISPLIT= 8
ATOM -2: X= 0.6950000 Y= 0.6950000 Z= 0.0000000
ATOM -2: X= 0.8050000 Y= 0.1950000 Z= 0.5000000
ATOM -2: X= 0.1950000 Y= 0.8050000 Z= 0.5000000
Oxygen NPT= 781 R0=.000017913 RMT=1.60000000 Z: 8.0
LOCAL ROT MATRIX: 0.0000000 -.7071068 0.7071068
0.0000000 0.7071068 0.7071068
1.0000000 0.0000000 0.0000000
16 SYMMETRY OPERATIONS: 11
1 0 0 0.00 12
0 1 0 0.00 13
0 0 1 0.00 14
1 15
1 0 0 0.00
0 1 0 0.00
0 0-1 0.00
2
........
15
0 1 0 0.50
-1 0 0 0.50
0 0 1 0.50
16
------------------ bottom of file ---------------------------
Interpretive comments on this file are as follows.
|
| lattice type | as defined in table 4.4. For definitions of the triclinic lattice see SRC_nn/dirlat.f | |
| NAT | number of inequivalent atoms in the unit cell |
| RELA | fully relativistic core and scalar relativistic valence | |
| NREL | non-relativistic calculation |
| a, b, c | unit cell parameters (in a.u., 1 a.u. = 0.529177 Å). In face- or body-centered structures the non-primitive (cubic) lattice constant, for rhombohedral (R) lattices the hexagonal lattice constants must be specified. (The following may help you to convert between hexagonal and rhombohedral specifications: | |
|
|
||
|
|
||
| and (for fcc-like lattices)
|
||
|
|
angles between unit axis (if omitted,
|
| atom-index | running index for inequivalent atoms | |
| positive in case of cubic symmetry | ||
| negative for non-cubic symmetry | ||
| this is set automatically using symmetry | ||
| x,y,z | position of atom in internal units,
i.e. as positive fractions of unit cell
parameters. ( |
|
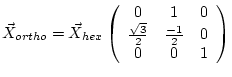 |
||
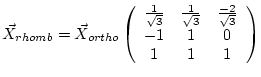 |
| multiplicity | number of equivalent atoms of this kind | |
| isplit | this is just an output-option and is used to specify the decomposition of the lm-like charges into irreducible representations, useful for interpretation in case.qtl). This parameter is automatically set by symmetry: | |
| 0 | no split of l-like charge | |
| 1 | p-z, (p-x, p-y) e.g.:hcp | |
| 2 | e-g, t-2g of d-electrons e.g.:cubic | |
| 3 | d-z2, (d-xy,d-x2y2), (d-xz,dyz) e.g.:hcp | |
| 4 | combining option 1 and 3 e.g.:hcp | |
| 5 | all d symmetries separate | |
| 6 | all p symmetries separate | |
| 8 | combining option 5 and 6 | |
| -2 | d-z2, d-x2y2, d-xy, (d-xz,d-yz) | |
| 88 | split |
|
| 99 | calculate cross-terms (for telnes) |
| name of atom | Use the chemical symbol. Positions 3-10 for further labeling of nonequivalent atoms (use a number in position 3) | |
| NPT | number of radial mesh points (381 gives a good mesh
for LDA calculations, but for GGA twice as many points
are recommended; always use an odd number of
mesh points!)
the radial mesh is given on a
logarithmic scale:
|
|
| R0 | first radial mesh point (typically between 0.0005 and 0.00005, smaller for heavy elements, bigger for light ones; a struct-file generated by w2web will have proper R0 values.) | |
| RMT | atomic sphere radius (muffin-tin radius), can easily be estimated after running nn (see 6.1) and are set automatically with setrmt_lapw see 5.2.6). The following guidelines will be given here: Choose spheres as large as possible as this will save MUCH computer time. But: Use identical radii within a series of calculations (i.e. when you want to compare total energies) -- therefore consider first how close the atoms may possibly come later on (volume or geometry optimization); do NOT make the spheres too different (even when the geometry would permit it), instead use the largest spheres for f-electron atoms, 10-20 % smaller ones for d-elements and again 10-20 % smaller for sp-elements; H is a special case, you may choose it much smaller (e.g. 0.6 and 1.2 for H and C) and systems containing H need a much smaller RKMAX value (3-5) in case.in1. | |
| Z | atomic number |
| ROTLOC | local rotation matrix (always in an orthogonal coordinate system). Transforms the global coordinate system (of the unit cell) into the local at the given atomic site as required by point group symmetry (see in the INPUT-Section 7.5.3 of LAPW2). SYMMETRY calculates the point group symmetry and determines ROTLOC automatically. Note, that a proper ROTLOC is required, if the LM values generated by SYMMETRY are used. A more detailed description with several examples is given in the appendix A and sec. 10.3 |
| nsym | number of symmetry operations of space group (see International Tables of Crystallography 64) | |
| If nsym is set to zero, the symmetry operations will be generated automatically by SYMMETRY. |
| matrix | matrix representation of (space group) symmetry operation | |
| tau | non-primitive translation vector |
During the self-consistent field (SCF) cycle the essential data are appended to the file case.scf in order to generate a summary of previous iterations. For an easier retrieval of certain quantities the essential lines are labeled with :LABEL:, which can be used to monitor these quantities during self-consistency as explained below. The most important :LABELs are
| :ENE | total energy (Ry) |
| :DIS | charge distance between last 2 iterations (
|
| :FER | Fermi energy |
| :FORxx | force on atom xx in mRy/bohr (in the local (for each atom) carthesian coordinate system) |
| :FGLxx | force on atom xx in mRy/bohr (in the global coordinate system of the unit cell (in the same way as the atomic positions are specified)) |
| :DTOxx | total difference charge density for atom xx between last 2 iterations |
| :CTOxx | total charge in sphere xx (mixed after MIXER) |
| :NTOxx | total charge in sphere xx (new (not mixed) from LAPW2+LCORE) |
| :QTLxx | partial charges in sphere xx |
| :EPLxx | l-like partial charges and ``mean energies'' in lower (semicore) energy window for atom xx. Used as energy parameters in case.in1 for next iteration |
| :EPHxx | l-like partial charges and ``mean energies'' in higher (valence) energy window for atom xx. Used as energy parameters in case.in1 for next iteration |
| :EFGxx | Electric field gradient (EFG) |
| :ETAxx | Asymmetry parameter of EFG for atom xx |
| :RTOxx | Density for atom xx at the nucleus (first radial mesh point) |
| :VZERO | Gives the total, Coulomb and xc-potential at z=0 and z=0.5 (meaningfull only for slab calculations) |
To check to which type of calculation a scf file corresponds use:
| :POT | Exchange-correlation potential used in this calculation |
| :LAT | Lattice parameters in this calculation |
| :VOL | Volume of the unit cell |
| :POSxx | Atomic positions for atom xx (as in case.struct) |
| :RKM | Actual matrix size and resulting RKmax |
| :NEC | normalization check of electronic charge densities. If a significant amount of electrons is missing, one might have core states, whose charge density is not completely confined within the respective atomic sphere. In such a case the corresponding states should be treated as band states (using LOs). |
For spin-polarized calculations:
| :MMTOT | Total spin magnetic moment/cell |
| :MMIxx | Spin magnetic moment of atom xx. Note, that this value depends on RMT. |
| :CUPxx | spin-up charge (mixed) in sphere xx |
| :CDNxx | spin-dn charge (mixed) in sphere xx |
| :NUPxx | spin-up charge (new, from lapw2+lcore) in sphere xx |
| :NDNxx | spin-dn charge (new, from lapw2+lcore) in sphere xx |
| :ORBxx | Orbital magnetic moment of atom xx (needs SO calculations and LAPWDM). |
| :HFFxx | Hyperfine field of atom xx (in kGauss). |
One can monitor the energy eigenvalues (listed for the first k-point only), the Fermi-energy or the total energy. Often the electronic charges per atom reflect the convergence. Charge transfer between the various atomic spheres is a typical process during the SCF cycles: large oscillations should be avoided by using a smaller mixing parameter; monotonic changes in one direction suggest a larger mixing parameter.
In spin-polarized calculations the magnetic moment per atomic site is an additional crucial quantity which could be used as convergence criterion.
If a system has electric field gradients and one is interested in that quantity, one should monitor the EFGs, because these are very sensitive quantities.
It is best to monitor several quantities, because often one quantity is converged, while another still changes from iteration to iteration. The script run_lapw has three different convergence criteria built in, namely the total energy, the atomic forces and the charge distance (see 5.1.2, 5.1.3).
We recommend the use of UNIX commands like :
grep :ENE case.scf or use ``Analysis'' from w2web
for monitoring such quantities.
You may define an alias for this (see sec. 11.2), and a csh-script grepline_lapw is also available to get a quantity from several scf-files simultaneously (sec. 5.2.16 and 5.3).
The WIEN2k package consists of several independent programs which are linked via C-SHELL SCRIPTS described below.
The flow and usage of the different programs is illustrated in the following diagram (Fig. 4.2):
The initialization consists of running a series of small auxiliary programs, which generates the inputs for the main programs. One starts in the respective case/ subdirectory and defines the structure in case.struct (see 4.3). The initialization can be invoked by the script init_lapw (see sec. 3.7 and 5.1.2), and consists of running:
Then a self-consistency cycle is initiated and repeated until convergence criteria are met (see 3.8 and 5.1.3). This cycle can be invoked with a script run_lapw, and consists of the following steps:
In many cases it is desirable to distinguish three types of electronic
states, namely core, semi-core and valence states. For example
titanium has core (![]() ,
, ![]() ,
, ![]() ), semi-core (
), semi-core (![]() ,
, ![]() ) and
valence (
) and
valence (![]() ,
, ![]() ,
, ![]() ) states. In our
definition core states are only those
whose charge is entirely confined inside the corresponding atomic
sphere. They are deep in energy, e.g., more than 7-10 Ry below the
Fermi energy. Semi-core states lie high enough in energy
(between about 1 and 7 Ry below the Fermi energy), so that their
charge is no longer completely confined inside the atomic sphere, but has a few percent
outside the sphere. Valence states are energetically the
highest (occupied) states and always have a significant amount of
charge outside the spheres.
) states. In our
definition core states are only those
whose charge is entirely confined inside the corresponding atomic
sphere. They are deep in energy, e.g., more than 7-10 Ry below the
Fermi energy. Semi-core states lie high enough in energy
(between about 1 and 7 Ry below the Fermi energy), so that their
charge is no longer completely confined inside the atomic sphere, but has a few percent
outside the sphere. Valence states are energetically the
highest (occupied) states and always have a significant amount of
charge outside the spheres.
The energy cut-off specified in lstart during init_lapw (usually -6.0 Ry) defines the separation into core- and band-states (the latter contain both, semicore and valence). If a system has atoms with semi-core states, then the best way to treat them is with ``local orbitals``, an extension of the usual LAPW basis. An input for such a basis set will be generated automatically. (Additional LOs can also be used for valence states which have a strong variation of their radial wavefunctions with energy (e.g. d states in TM compounds) to improve the quality of the basis set, i.e. to go beyond the simple linearization).
For magnetic systems spin-polarized calculations can be performed. In such a case some steps are done for spin-up and spin-down electrons separately and the script runsp_lapw consists of the following steps:
The use of spin-polarized calculations is illustrated for fcc Ni (section 10.2), one of the test cases provided in the WIEN2k package.
Using the script runfsm_lapw -m XX it is possible to constrain the total spin magnetic moment per unit cell to a fixed value XX and thus force a particular ferromagnetic solution (which may not correspond to the equillibrium). This is particularly useful for systems with several metastable (non-) magnetic solutions, where conventional spin-polarized calculation would not converge or the solution may depend on the starting density. Additional SO-interaction is not supported.
Please note, that once runfsm_lapw has finished, only case.vectordn is ok, but case.vectorup is NOT the proper up-spin vector and MUST NOT be used for the calculations of QTLs (and DOS). It must be regenerated by x lapw1 -up (see also the comments for iterative diagonalization in section 5.2.18).
Several considerations are necessary, when you want to perform an AFM calculation. Please have also a look into $WIENROOT/SRC_afminput/afminput_test.
runafm_lapw saves you more than a factor of 2 in in computer time, since only spin-up is calculated and in addition the scf-convergence may be MUCH faster. It works also with LDA+U (case.dmatup/dn are also copied), but does NOT work with Hybrid-DFT nor spin-orbit coupling, since this requires the presence of both vector files in the LAPWSO step.
You can add spin-orbit interaction in LAPWSO (called directly after LAPW1)
using a second-variational method with
the scalar-relativistic orbitals (from LAPW1) as basis. The number of
eigenvalues will double since SO couples spin-up and dn states, so they are
no longer separable. In addition, LOs with a ``![]() ''
radial basis can be added. (Kunes et al. 2001)
''
radial basis can be added. (Kunes et al. 2001)
To assist with the generation of the necessary input files and possible changes in symmetry, a script initso_lapw exists. For non-spinpolarized cases nothing particular must be taken into account and SO can be easily applied by running run_lapw -so. It will automatically use the complex version of LAPW2.
However, for spin-polarized cases, the SO interaction may change (lower) the symmetry depending on how you choose the direction of magnetization and care must be taken to get a proper setup. initso_lapw together with symmetso generates the proper symmetry.
Just a few hints what can happen:
The recommended way to include SO in the calculations is to run a regular scf calculation first, save the results, initialize SO and run another scf cycle including SO:
For spin-polarized systems you may want to add the ``-dm'' switch to calculate also the orbital magnetic moment.
To use these features you need to create input-files for LAPWDM and ORB (case.indm, case.inorb). You may copy a template from SRC_templates, but must modify it according to your needs. In particular you must select for which atoms and which orbitals (usually d-Orbitals of late transition metal atoms or f-orbitals for 4f/5f atoms) you want to add such a potential and also choose the proper U and J values for them. Once this is done, you can include this using the -orb switch. The density matrix (case.dmatup/dn) will be calculated after lapw2 in lapwdm, it will be mixed in mixer (consistently with the ``regular'' charge density) and the orbital dependend potentials will be calculated on orb (after lapw0). Note, you must run spin-polarized in order to use orbital potentials.
If you want to force a non-magnetic solution you can constrain the spin-polarization to zero using runsp_c_lapw.
Without SO, case.vorbup/dn will be considered in LAPW1(c). With SO, it will be applied in LAPWSO (and allows coupling of nondiagonal spin-terms).
Examples of implemented functionals include:
![\begin{displaymath}
E_{xc}^{\textrm{LDA-HF}}[\rho] = E_{xc}^{\textrm{LDA}}[\rho]...
...{\textrm{corr}}] -
E_{xc}^{\textrm{LDA}}[\rho_{\textrm{corr}}]
\end{displaymath}](img124.png)
![\begin{displaymath}
E_{xc}^{{\textrm{LDA-Fock-}}\alpha}[\rho] = E_{xc}^{\textrm{...
...rm{corr}}] -
E_{x}^{\textrm{LDA}}[\rho_{\textrm{corr}}]\right)
\end{displaymath}](img125.png)
![\begin{displaymath}
E_{xc}^{\textrm{PBE-Fock-}\alpha}[\rho] = E_{xc}^{\textrm{PB...
...rm{corr}}] -
E_{x}^{\textrm{PBE}}[\rho_{\textrm{corr}}]\right)
\end{displaymath}](img126.png)
![\begin{eqnarray*}
E_{xc}^{\textrm{B3PW91}}[\rho] & = & E_{xc}^{\textrm{LDA}}[\rh...
...(E_{c}^{\textrm{PW91}}[\rho] -
E_{c}^{\textrm{LDA}}[\rho]\right)
\end{eqnarray*}](img131.png)
In addition to the input files which are necessary for an usual LDA or GGA calculation, the input file case.ineece is necessary to start a calculation. You may copy a template from SRC_templates, but must modify it according to your needs. In particular you must select for which atoms and which orbitals (usually d-Orbitals of late transition metal atoms or f-orbitals for 4f/5f atoms) you want to add such a potential and which type of functional you want to use.
A sample input for calculations with exact exchange is given below.
------------------ top of file: case.ineece ----------- -9.0 2 emin, natorb 1 1 2 1st atom index, nlorb, lorb 2 1 2 2nd atom index, nlorb, lorb HYBR HYBR / EECE mode 0.25 fraction of exact exchange ------------------ bottom of file ---------------------
Interpretive comments on this file are as follows:
line 1: free format
emin, natom
| emin | lower energy cutoff, to be selected so that the energy of correlated states is larger than emin | |
| natorb | number of atoms for which the exact exchange is calculated |
| iatom | index of atom in struct file | |
| nlorb | number of orbital moments for which exact exchange shall be calculated | |
| lorb | orbital numbers (repeated nlorb-times) |
line 3: free format
mode
| HYBR | means that LDA/GGA exchange will be replaced by exact exchange | |
| EECE | means that LDA/GGA exchange-correlation will be replaced by exact exchange |
| alpha | This is the fraction of Hartree-Fock exchange (between 0 and 1) |
As with LDA+![]() , hybrid functionals can be used only for spin-polarized
calculations (runsp_lapw with the switch -eece).
runsp_lapw will internally call runeece_lapw, which
will create all necessary additional input files (it requires a case.in0 file
including the optional IFFT line as generated by init_lapw):
case.indm (case.indmc), case.inorb, case.in0eece, case.in2eece
(case.in2ceece)
and once this is done, calculates in a series of lapw2/lapwdm/lapw0/orb
calculations the corresponding orbital dependend potentials.
, hybrid functionals can be used only for spin-polarized
calculations (runsp_lapw with the switch -eece).
runsp_lapw will internally call runeece_lapw, which
will create all necessary additional input files (it requires a case.in0 file
including the optional IFFT line as generated by init_lapw):
case.indm (case.indmc), case.inorb, case.in0eece, case.in2eece
(case.in2ceece)
and once this is done, calculates in a series of lapw2/lapwdm/lapw0/orb
calculations the corresponding orbital dependend potentials.
The modified Becke-Johnson exchange potential + LDA-correlation (Tran and Blaha
2009) allows the calculation of band gaps with an accuracy similar to very
expensive GW calculations. It is a local approximation to an atomic
``exact-exchange''-potential and a screening term. This is just a XC-potential, not a XC-energy
functional, thus ![]() is taken from LSDA and the forces cannot be used with
this option.
is taken from LSDA and the forces cannot be used with
this option.
We recommend the following steps to perform such calculations:
In most cases MSEC1 mixing will lead to convergence problems of the scf cycle. One should switch in case.inm to PRATT mixing, first using a smaller mixing factor (eg. 0.2), later increasing it to 0.50.